The goal of our basic research endeavors focuses primarily on developing effective gene and cell therapies for urea cycle disorders. Our desire is to develop new therapies that would replace liver transplantation for single enzyme metabolic deficiencies. Specifically, the lab’s efforts have been to develop such approaches in neonatal or in utero models of human disease focusing on nitrogen metabolism disorders: primarily arginase deficiency, and more recently also on carbamoyl phosphate synthetase (CPS1) deficiency. Both of these urea cycle defects demonstrate intellectual disabilities and growth retardation in humans; the cause and mechanism of brain injury appears to be different with CPS1 deficiency caused by hyperammonemia, often resulting in early mortality, while in arginase deficiency elevated ammonia is uncommon and the disease is relentless and progressive. There is no completely effective therapy for these disorders and we are developing gene-and cell based approaches for these. Furthermore, as part of this endeavor, we have directed considerable effort to try to understand the underlying neuropathology that occurs in arginase deficiency with hyperargininemia.
We have four active research directions:
- Developing Gene- and Cell-Based Hepatic Therapies
We aim to develop new gene- and cell-based therapies for treating metabolic disorders of the liver. Our strategies have included the development of iPSCs from patients with arginase deficiency, CRISPR/Cas9-based genomic correction, and development of hepatocyte-like cells for transplantation, AAV-based approaches for arginase deficiency and a unique dual vector system for CPS1 deficiency; and mRNA with lipid nanoparticle administration. Studies of enzyme expression levels, RNA, and functional activity using murine models of these disorders aims to demonstrate a proof-of-principle of these therapies. - Studying the Developing Nervous System with Hyperargininemia (Arginase Deficiency)
We aim to understand the mechanism of brain injury caused by hyperargininemia and determine the toxin causing this injury. We have recently determined that marked dysmyelination occurs in this disorder; our data support a change in the classification of this disorder to now also its inclusion as a leukodystrophy and not solely a disorder of nitrogen metabolism. Based on this and other recent findings we intend to both identify the molecules that result in the presumed oligodendrocyte dysfunction that is occurring and to further understand the role that neuronal and glial arginase plays in the CNS.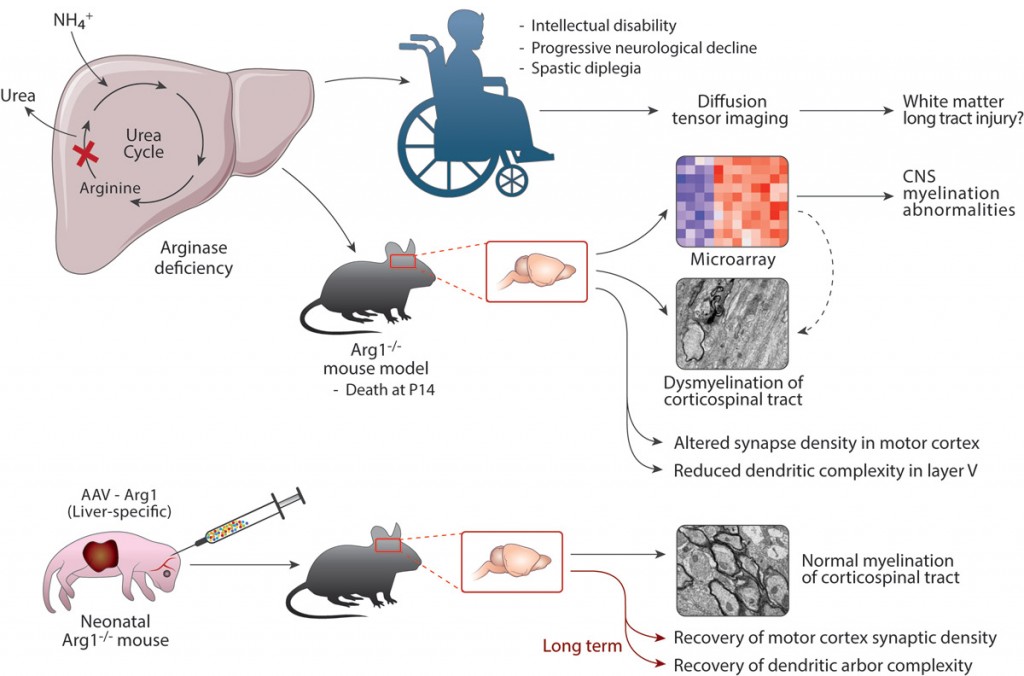
- Developing Gene Therapy Approaches for Carbamoyl Phosphate Synthetase 1 Deficiency
We aim to develop a gene therapy approach for Carbamoyl Phosphate Synthetase 1 deficiency. This proximal urea cycle disorder results in the early and rapid rise of ammonia shortly after birth. If not recognized early, children who survive and typically afflicted with significant brain injury. At present, the only therapy if liver transplantation in those that survive the first episode of hyperammonemia. This disorder if particularly challenged by, until recently, the lack of a viable mouse model of the disorder, the large amount of this mitochondrial protein necessary to treat the disorder (CPS1 is ~20% of matrix mitochondrial protein), and the large size of the CPS1 cDNA (4.521 kb). The lab has been developing strategies to overcome these limitations. 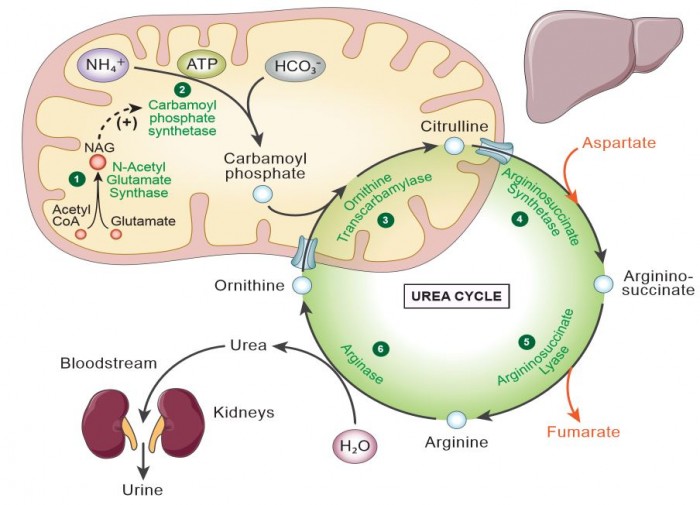 Developing a Gene Therapy Approach for Guanidinoacetate Methyltransferase Deficiency and Understanding the mechanism of Guanidinoacetate-mediated injury to the Brain
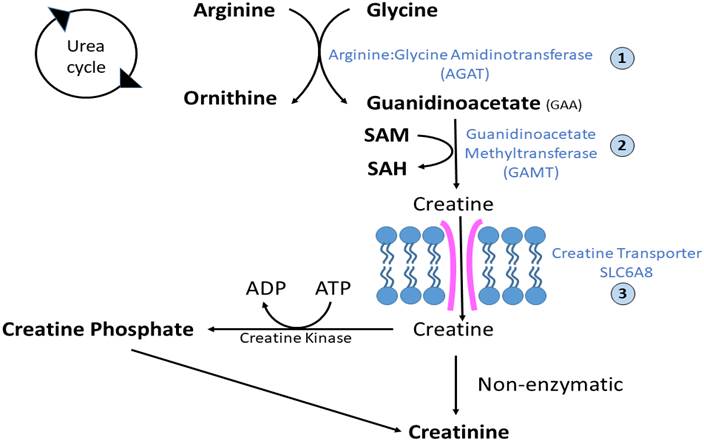
Developing a Gene Therapy Approach for Guanidinoacetate Methyltransferase Deficiency and Understanding the mechanism of Guanidinoacetate-mediated injury to the Brain
We aim to develop a gene therapy approach for a creatine deficiency disorder, Guanidinoacetate methyltransferase (GAMT) deficiency. The deficiency often results in speech abnormalities, seizures, movement disorders, and an autistic-like behavioral phenotype. Our group has been actively developing a gene therapy approach to treat this condition. At the same time, we are working to develop a better understanding of the effect of the abnormal metabolite guanidinoacetate (GAA) and its effect of the function of neurons in the CNS.